
| Al primer inventor pertenece todo el mérito | Píndaro |
Un plásmido es una pequeña molécula circular de ADN que se encuentra principalmente en el citoplasma de muchas bacterias, aunque también pero en menor medida en organismos eucariotas, como las levaduras. Una de las características distintivas que poseen los plásmidos es que es independiente del genoma del organismo que lo lleva consigo. Además, cuenta con la capacidad de replicarse por sí solo, es decir, de manera autónoma e independiente al ADN del nucleoide. En una célula pueden encontrarse desde uno a cientos de plásmidos, y cada uno puede poseer de miles a decenas de miles de pares de bases.
Los plásmidos portan genes que contienen información para producir proteínas que proporcionan resistencia a antibióticos a la bacteria o enzimas capaces de degradar ciertas sustancias. Esto es importante ya que los plásmidos son transferibles de unas bacterias a otras, transmitiéndose también dichas capacidades. Esta transferencia se lleva a cabo por conjugación; una bacteria emite una prolongación de su citoplasma, un puente citoplasmático, que contacta con otra bacteria y a través del cual pasa de una a otra. Estas moléculas tienen mucha utilidad en los laboratorios y en la manipulación genética para la obtención de transgénicos, empleándose como vectores de transferencia.
Por otro lado, los enzimas de restricción, son proteínas capaces de reconocer ciertas secuencias de nucleótidos muy específicas en el ADN y de cortar (hidrolizar) los enlaces fosfodiéster que los unen. El lugar sobre el que actúan las proteínas son las dianas de restricción. Para las bacterias, estos enzimas son una defensa contra virus, ya que cortan su material genético cuando estos las atacan.
Los enzimas cortan en unas secuencias denominadas palindrómicas, aquellas que se leen de la misma forma de izquierda a derecha que de derecha a izquierda; se leen igual del extremo 5' al 3' que del 3' al 5'. Cuando cortan en un ADN, en las hebras resultan extremos que son cohesivos y complementarios, lo que facilita que puedan enlazarse entre sí con ayuda de otros enzimas conocidos como ADN ligasas. También pueden originar extremos romos, cuando cortan cadenas de material genético por el mismo lugar.
Mediante enzimas de restricción, se efectúan cortes en un ADN con el fin de extraer un gen de interés. Posteriormente, se realiza otro corte con el mismo enzima en un plásmido, que gracias a la complementariedad de los extremos a los que se ha dado lugar en ambas cadenas de material genético y a las ADN ligasas, el gen de interés se une a este dando lugar a un ADN recombinante. Así, sometiendo a descargas eléctricas a una bacteria, que incrementan la permeabilidad de su membrana, el plásmido se introduce dentro de esta, de tal manera que la célula contará con un gen extraño que le conferirá una capacidad con la que no contaba anteriormente.
Después del cultivo de bacterias fluorescentes, realizamos una digestión doble de ADN con enzimas de restricción además de una electroforesis. Analizamos los resultados, comprobando la realización de los cortes hechos por los enzimas.
After the cultivation of fluorescent bacteria, we perform a double digestion of DNA with restriction enzymes as well as an electrophoresis. We analyze the results, checking the making of the cuts made by the enzymes.
El material utilizado para realizar el trabajo de laboratorio fue: 5 cultivos de bacterias (modificadas genéticamente, fluorescentes y no modificadas genéticamente, no fluorescentes), el asa de siembra, un mechero de alcohol, tubos eppendorf, micropipetas, buffer P1 (con RNasa), buffer P2 (con SDS y NaOH), buffer P3, buffer TE, centrifugadora, SDS, alcoholes (etanol e isopropanol), cámara de electroforesis, gel green, azul de bromofenol (con glicerina/glicerol), enzimas de restricción (Eco RI, Hind III y Bam HI), agua, baño maría, plásmidos, gel 0,7% agarosa, tampón TBE y peine.
Inicialmente, introducimos utilizando la micropipeta 200 µl del buffer P1 en cada uno de los cuatro tubos eppendorf. Comenzando con la muestra que contiene la proteína de la fluorescencia, calentamos el asa de siembra hasta que alcanza un color rojo, para luego ser arrastrada con cuidado sobre los cultivos, sin retirar el agar junto a las bacterias y se introduce la muestra recogida en un tubo eppendorf que contiene el buffer P1. Seguimos este procedimiento con los otros cultivos de bacterias, hasta que cada tubo contenga una muestra. Lo siguiente es mezclar el tampón con las bacterias, para ello, las mezclamos bien utilizando una micropipeta mediante succiones repetidas y dejar incubar unos minutos en hielo.
Pasado el tiempo, introducimos 200 µl de buffer P2 que contiene SDS (detergente que rompe las membranas celulares) en cada muestra y las agitamos, hasta que alcancen una consistencia de clara de huevo. Se incuba durante 2 minutos a temperatura ambiente. Tras esto, añadimos 200 µl de buffer P3 (acetato potásico), el cual favorece que precipite el ADN sin proteínas, se deja otros dos minutos incubando a temperatura ambiente. Además, dejamos las muestras en la centrifugadora durante 10 minutos a 7000 revoluciones. Después, extraemos el líquido sobrante con cuidado de no extraer la muestra que se encuentra en el fondo.
A continuación, se introducen 500 µl de alcohol (isopropanol), produce que precipite el ADN, ya que es insoluble en alcohol. Se agitan y se colocan las muestras en la centrifugadora otros 10 minutos. Pasado este tiempo, en el fondo del tubo se tiene que ver un pequeño sedimento de aspecto blanquecino del tamaño de una cabeza de alfiler que es el DNA plasmídico. Se descarta el sobrenadante por decantación y posteriormente, añadimos 500 µl de etanol frío al 70% para que la precipitación se estabilice, centrifugar 1 minuto y descartar el sobrenadante. Después, centrifugar 5 segundos, para que caigan las gotas de etanol y poder quitarlo con una micropipeta, luego dejar secar unos minutos. Para finalizar, se añaden 40 µl de TE (mantiene el pH en 8), disolviéndolo y se introduce en el congelador hasta que vayan a ser utilizadas las muestras.
Antes de digerir los plásmidos, comprobamos que realmente los hemos extraído, para ello hacemos un gel de agarosa y mediante electroforesis, comprobamos que estos realmente han sido extraídos.
Para la digestión es necesario introducir en cada tubo eppendorf 2 µl de plásmido, 1 µl de enzima (añadimos otro si la digestión es doble), 3 µl de buffer y 23 µl de agua. Se mezclan y se dejan durante una hora y media en el baño maría a 37ºC (usando un flotador).
La observación de los resultados se realiza mediante electroforesis horizontal, técnica utilizada para separar sustancias mediante el uso de la corriente eléctrica, dependiendo del tamaño y carga eléctrica de la muestra va a desplazarse de una forma diferente con la corriente.
Partiendo de la cámara de electroforesis se retira el peine que se encuentra sobre el gel de agarosa, dejando 7 pocillos en éste. Se rellena con 30 ml del tampón TBE la cámara. Se utiliza azul de bromofenol para marcar 7 pocillos y poder insertar mejor los plásmidos. Tenemos 6 muestras, las 3 primeras son el mismo plásmido que no contiene la proteína fluorescente y las otras sí. Las calles 2 y 5 fueron digeridas con los enzimas Eco RI y Hind III, las calles 3 y 6 con Eco RI y Bam HI; y calles 4 y 7 con Hind III y Bam HI
Lo siguiente es rellenar 6 de los pocillos con 20 µl de las diferentes muestras. Dejando uno de los pocillos para el Marker III (marcador de peso molecular). Para acabar, conectamos los polos de la fuente de alimentación. Se comprueba que funciona correctamente si comienzan a salir burbujas a ambos lados, lo dejamos durante una hora funcionando. La fuente de alimentación es de 48 V.
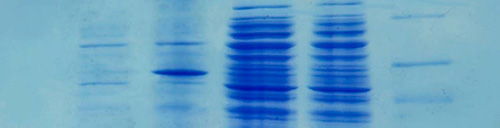
Antes de realizar las digestiones de los plásmidos para su caracterización enzimática, se comprobó mediante una electroforesis que el ADN se había extraído con éxito. Al llevarla a cabo, los plásmidos avanzaron por el gel de agarosa, confirmando que, efectivamente, se había conseguido aislarlos como lo confirman las bandas que se visualizan en el gel (figura 1).
Los resultados obtenidos en las digestiones dobles con los enzimas de restricción se muestran en la figura 2.
Los plásmidos que no presentan el gen que emite fluorescencia, esto es, las calles 2, 3 y 4, muestran una única banda visible tras haber realizado la electroforesis de los productos de las digestiones. Esta banda se corresponde con un fragmento en las tres muestras de un tamaño aproximado de 5,1 Kb (kilobases), como nos indica el marcador de peso molecular.
Los plásmidos portadores del gen que aporta fluorescencia, es decir, las calles 5, 6 y 7 muestran, por lo general, dos bandas visibles en el gel de electroforesis. En el caso de la calle 5 (cortado con los enzimas EcoRI/HindIII), se observan con claridad dos bandas, una que representa un fragmento de ADN de alrededor de 4,3 kb y otro de aproximadamente 0,93 kb. En la calle 6 que contiene el plásmido cortado con los enzimas EcoRI/BamHI observamos un solo fragmento de unas 4,3 kb. Finalmente, en la calle 7 (enzimas HindIII/BamHI) observamos de nuevo dos bandas, una por debajo de las 4,3 kb y otra de un valor cercano a 0,8 kb que no se puede diferenciar en el marcador.
La única banda que se observa tras las digestiones en el plásmido no fluorescente (calles 2 a 4, figura 2) se debe a que las dianas de restricción, es decir, las secuencias en las que los enzimas de restricción cortan en la cadena de material genético, están muy próximas una de la otra. De esta manera, en esos tres casos se da lugar a un fragmento largo de ADN, representado en la banda visible, y a otro muy pequeño, cuya visualización no es posible al no contar con los suficientes pares de bases como para verse reflejado en el gel. El fragmento pequeño de plásmido del que no se tiene constancia según lo que se puede ver en la electroforesis debe de tener un tamaño menor de 0,8 Kb; ha de estar constituido por una cantidad inferior a 800 pares de bases. En concordancia con los resultados observados, un posible mapa de restricción del plásmido es el que se muestra en la figura 3.
Como se puede ver en la figura 2, los plásmidos con el gen fluorescente muestran dos bandas, excepto en la calle 6, correspondiente a la digestión con EcoRI/BamHI, que sólo se visualiza una banda. En este caso, deducimos que la concentración de plásmido no es suficiente para hacer visible una banda de tan pequeño tamaño. Atendiendo a los resultados obtenidos mediante las dobles digestiones del plásmido, se propone un posible mapa de restricción que se muestra en la figura 4.
| Figura 1: Electroforesis en gel de agarosa. 1 Plásmido no modificado; 2, 3 y 4. Plásmidos modificados genéticamente; 5 Marcador de peso molecular (Marker III). |  |
 |
Figura 2: Electroforesis de digestiones en gel de agarosa. 1 Marcador de peso molecular (Marker III); 2 Digestión Eco RI/Hind III; 3 Digestión Eco RI/Bam HI; 4 Digestión Hind III/Bam HI; 5 Digestión Eco RI/Hind III; 6 Digestión Eco RI/Bam HI; 7 Digestión Hind III/Bam HI. |
| Figura 3: Posibles dianas de restricción en el plásmido no fluorescente para los enzimas Eco RI, Hind III y Bam HI. |  |
 |
Figura 4: Posibles dianas de restricción en el plásmido fluorescente para los enzimas Eco RI, Hind III y Bam HI. |
- https://www.sabermas.umich.mx/archivo/articulos/283-numero-33/510-plasmidos bacterianos.html.
- https://es.khanacademy.org/science/biology/biotech-dna-technology/dna-cloning-tutorial/a/restriction-enzymes-dna-ligase.
